Key contribution: Propose a statistical method for cross-population fine-mapping common causal SNPs (XMAP) by leveraging genetic diversity (different LD structures) and accounting for confounding bias which addresses the challenges of:
Strong linkage disequilibrium among variants can limit the statistical power and resolution of fine-mapping. [Solved by LDSC and SuSiE]
Computationally expensive to simultaneously search for multiple causal variants. [Solved by SuSiE]
The confounding bias hidden in GWAS summary statistics can produce spurious signals. [Adjusting by the inflation factor c]
It also integrates the polygenic component ϕ to capture the genetic background effects.
It can be integrated with single cell data to identify trait-relevant cell populations at the single cell resolution.
Advantages over existing methods: Greater statistical power, better calibrate of false positive rate, and substantially higher computational efficiency for identifying multiple causal signals.
Input: GWAS datasets {y1,X1} and {y2,X2} from two different populations, where y1∈Rn1 and y2∈Rn2 are phenotype vectors, X1∈Rn1×p and X2∈Rn2×p are genotype matrices, p is the number of SNPs in the locus of interest, and n1 and n2 are the GWAS sample sizes of populations 1 and 2 , respectively. Assume that the columns of X1 and X2 have been standardized to have zero mean and unit variance.
Model:
Linear models to relate genotypes and phenotypes: y1=X1b1+X1ϕ1+e1,y2=X2b2+X2ϕ2+e2,
where b1∈Rp and b2∈Rp are sparse vectors of causal effects with major impact on phenotypes, ϕ1=[ϕ11,ϕ12,…,ϕ1p]T∈Rp and ϕ2=[ϕ21,ϕ22,…,ϕ2p]T∈Rp are dense vectors capturing the polygenic effects, and e1∼N(0,σe12In1) and e2∼N(0,σe22In2) are vectors of independent noises from populations 1 and 2 , respectively.
Decomposition of the causal genetic effects b1 and b2 into K 'single effects': y1=X1k=1∑Kγkβ1k+X1ϕ1+e1,y2=X2k=1∑Kγkβ2k+X2ϕ2+e2,
where β1k and β2k are effect sizes of the k-th causal signal in populations one and two, respectively, γk=[γk1,…,γkp]T∈{0,1}p in which only one element is 1 and the rest are 0 with γkj=1 indicating the j-th variant is responsible for the k-th causal signal.
Probabilistic structures for the genetic effects: γk∼Mult(1,[1/p,…,1/p]T),[β1kβ2k]∼N(0,Σk), for k=1,…,K,[ϕ1jϕ2j]∼N(0,Ω), for j=1,…,p,
where Mult (1,[1/p,…,1/p]T) denotes the non-informative categorical distribution of class counts drawn with class probabilities given by 1/p for each SNP, N(0,Σk) and N(0,Ω) denote the multivariate normal distributions with mean 0 and covariance matrices Σk=[σk12σk122σk122σk22] and Ω=[ω1ω12ω12ω2], respectively.
Covariates and adjustment: [shows the equivalence of the model with and without covariates] y1=W1u1+X1k=1∑Kγkβ1k+X1Φ1+e1y2=W2u2+X2k=1∑Kγkβ2k+X2Φ2+e2
where W1∈Rn1×q1 and W2∈Rn2×q2 are the covariate matrices of populations 1 and 2, respectively, and u1∈Rq1 and u2∈Rq2 are corresponding vectors of covariate effects. To adjust the covariates, we first construct the projection matrices P1=I−W1(W1TW1)−1W1T and P2=I−W2(W2TW2)−1W2T. Then we multiply P1 on both sides of the first equation and P2 on both sides of the second equation in model (4). Through this projection, we can obtain a model without covariates: y1P=X1Pk=1∑Kγkβ1k+X1PΦ1+e1Py2P=X2Pk=1∑Kγkβ2k+X2Pϕ2+e2P
where y1P=P1y1,y2P=P2y2,X1P=P1X1,X2P=P2X2,e1P=P1e1, and e2P=P2e2. As we can observe, model (5) reduces to model (2). With this equivalence, we can work with model (2) without loss of generality.
XMAP Model for Summary-level Data
Input:
Summary-level GWAS data {b^1,s^1}={b^1j,s^1j}j=1,…,p and {b^2,s^2}={b^2j,s^2j}j=1,…,p obtained from simple linear regressions: b^1j=x1jTy1/x1jTx1j,b^2j=x2jTy2/x2jTx2j,s^1j=y1−x1jb^1j22/(n1x1jTx1j),s^2j=y2−x2jb^2j22/(n2x2jTx2j),
where x1j∈Rp and x2j∈Rp denote the j-th column of X1 and X2, respectively.
LD matrices R1={r1jl}∈Rp×p and R2={r2jl}∈Rp×p, where r1jl=E[x1jTx1l/n1] and r2jl=E[x2jTx2l/n2] denote the correlation between variants j and l in populations 1 and 2, respectively. The SNP correlation matrices R={R1,R2} can be estimated with genotypes either from subsets of GWAS samples or from population-matched reference panels.
Model:
Expectation of GWAS effect sizes conditional on b and ϕ: E[b^1∣b1,ϕ1]=R1k=1∑Kγkβ1k+R1ϕ1,E[b^2∣b2,ϕ2]=R2k=1∑Kγkβ2k+R2ϕ2
Note: E[b^∣…]=E[XTy/XTX∣…],XTX=1.
Distribution of b^: Assuming normal distributions for b^1 and b^2, we have: b^1∼N(R1k=1∑Kγkβ1k+R1Φ1,S^1R1S^1),b^2∼N(R2k=1∑Kγkβ2k+R2Φ2,S^2R2S^2).
where S^1∈Rp×p and S^2∈Rp×p are diagonal matrices with diagonal terms given as {S^1}jj=s^1j and {S^2}jj=s^2j for j=1,…,p. And S^1RS^1 and S^2RS^2 are the variances of ϵ^1=X^1Te^1 and ϵ^2=X^2Te^2, respectively.
Account for confounding bias: Modify (8) to account for the unadjusted confounding bias: b^1∼N(R1k=1∑Kγkβ1k+R1Φ1,c1S^1R1S^1),b^2∼N(R2k=1∑Kγkβ2k+R2Φ2,c2S^2R2S^2).
where c1 and c2 are LDSC intercepts that indicate the magnitude of inflation in GWAS effect sizes due to confounding bias. In the absence of confounding bias, the values of inflation constants c1 and c2 are close to one.
Algorithm and Parameter Estimation
Denote the collection of unknown parameters θ={Σ,Ω,c1,c2}, and the collections of latent variables ϕ={ϕ1,ϕ2},γ={γk}k=1,…,K and β={β1k,β2k}k=1,…,K. Obtain the parameter estimates θ and identify causal SNPs with the posterior: Pr(γ,β,Φ∣b^,s^,R;θ^)=Pr(b^∣s^,R;θ^)Pr(b^,γ,β,ϕ∣s^,R;θ^).
First step: Apply LDSC to estimate the parameters c1,c2, and Ω.
For Ω, the diagonal terms ω1 and ω2 are estimated with the per-SNP heritabilities of the corresponding populations using LDSC. The off-diagonal term ω12 is estimated by the per-SNP co-heritability obtained via bi-variate LDSC.
The inflation constants c1 and c2 are estimated by the intercepts of LDSC of the two populations.
Second step: Variational expectation-maximization (VEM) algorithm to estimate Σ.
Derive a lower bound of the logarithm of the marginal likelihood: logPr(b^∣s^,R;Ω^,c^1,c^2,Σ)≥γ∑∬q(γ,β,ϕ)logq(β,ϕ)Pr(b^,γ,β,ϕ∣s^,Ω^,c^1,c^2,Σ)dβdϕ=Eq[logPr(b^,γ,β,Φ∣s^,R;Ω^,c^1,c^2,Σ)−logq(γ,β,Φ)]≡Lq(Σ)
Factorizable formulation of the mean field variational approximation: q(γ,β,ϕ)=k=1∏Kq(b1k,b2k)q(ϕ)=k=1∏Kq(γk)q(β1k,β2k∣γk)q(ϕ),
where q(b1k,b2k)=q(γk)q(β1k,β2k∣γk) and q(ϕ) are the distributions of {b1k,b2k} and ϕ under the variational approximation, respectively.
E-step: Variational distributions at the t-th iteration are given as: q(γk∣Σ(t))=Mult(1,π~k),q([β1kβ2k]γkj=1,Σ(t))=N(μ~kj,Σ~kj),q([Φ1Φ2]Σ(t))=N(v~,Λ~),
where π~=[π~k1,…,π~kp]T∈[0,1]p,Σ~kj∈R2×2,μ~kj∈R2,Λ~∈R2p×2p, and v~∈R2p are variational parameters. The variational parameters are given as π~kjΣ~kjμ~kjΛ~v~=softmax(−log(p)+21logΣ~kj+21μ~kjTΣ~kj−1μ~kj),=[σ~kj,12σ~kj,22σ~kj,122σ~kj,22]=([c^1s^1j2r1j00c^2s^2j2r2j]+(Σk(t))−1)−1,=[μ~kj,1μ~kj,2]=Σ~kjc^1s^1j2b^1jc^2s^2j2b^2j−c^1s^1j2R1jT00c^2s^2j2R2jTk′=1∑Kμ~k′j⊗π~k′+v~,=([c^1s^1−1R1s^1−100c^2s^2−1R2s^2−1]+Ω^−1⊗Ip)−1,=Λ~[c^1s^1−2b^1c^2s^2−2b^2]−[c^1s^1−1R1s^1−100c^2s^2−1R2s^2−1](k=1∑Kμ~kj⊗π~k),
where softmax denotes the softmax function to make sure ∑j=1pπ~kj=1 and ⊗ is the Kronecker product. The lower bound (11) can be analytically evaluated as Lq(Σ∣Σ(t))=(k∑Kμ~kj⊗π~k+v~)T[c^1s^1−2b^1c^2s^2−2b^2]−21(k∑Kμ~kj⊗π~k+v~)T×[c^1s^1−1R1s^1−100c^2s^2−1R2s^2−1](k∑Kμ~kj⊗π~k+v~)−j∑p2c^1s^1j21r1jjk∑Kπ~kj(μ~kj,12+σ~kj,12)−j∑p2c^2s^b,2j21r2jjk∑Kπ~kj(μ~kj,22+σ~kj,22)+21k∑K((μ~kj⊗m~k)T[c^1s^1−1R1s^1−100c^2s^2−1R2s^2−1](μ~kj⊗π~k))−2p1k∑j∑Tr(Σk−1(Σ~kj+μ~kjμ~kjT))−2plog∣2πΩ^∣−21v~T(Ω^−1⊗Ip)v~−21Tr(([c^11S^1−1R1S^1−100c^21S^2−1R2S^2−1]+Ω^−1⊗Ip)Λ~)+j∑pk∑Kπ~kjlogp1−j∑pk∑Kπ~kjlogπ~kj+21j∑pk∑Kπ~kj(logΣ~kj−log∣Σk∣)+21log∣Λ~∣+ constant
where Tr(B) denotes the trace of the square matrix B, and the constant term does not involve Σ.
M-step: Solve ∂Σk∂Lq=0 to obtain the update equation of Σk: Σk(t+1)=j∑pπ~kj(μ~kjμ~kjT+Σ~kj)
Identification of Causal Variant and Construction of Credible Set (Output)
Posterior Inclusion Probability (PIP): The posterior inclusion probability of SNP j is computed as: PIPj=Pr(γkj=0 for some k∣b^,s^)=1−k=1∏K(1−π~kj)
where π~kj is the posterior probability that the k-th causal signal is contributed by the j-th SNP in equation (14). By controlling the FDR, we can prioritize the causal SNPs by computing the local FDR of SNP j as fdrj=1−PIPj.
Creditable set: The level-α credible set of a causal signal k, denoted as CS(k,α), is defined as the smallest set of SNPs with ∑j∈CS(k,α)PIPj≥α.
Conclusion
Comparison with other methods (including DAP-G, FINEMAP, SuSiE, SuSiE-inf, PAINTOR, MsCAVIAR, and SuSiEx in simulation study; SuSiE, SuSiE-inf and SuSiEx in real data analysis), shows that XMAP has three features:
It can better distinguish causal variants from a set of associated variants by leveraging different LD structures of genetically diverged populations.
By jointly modeling SNPs with putative causal effects and polygenic effects, XMAP allows a linear-time computational cost to identify multiple causal variants, even in the presence of an over-specified number of causal variants.
It further corrects confounding bias hidden in the GWAS summary statistics to reduce false positive findings and improve replication rates.
In particular, XMAP results can be effectively integrated with single-cell datasets to identify disease/trait-relevant cells.
Simulation Study
Data: The chosen of simulation data considers the following factors:
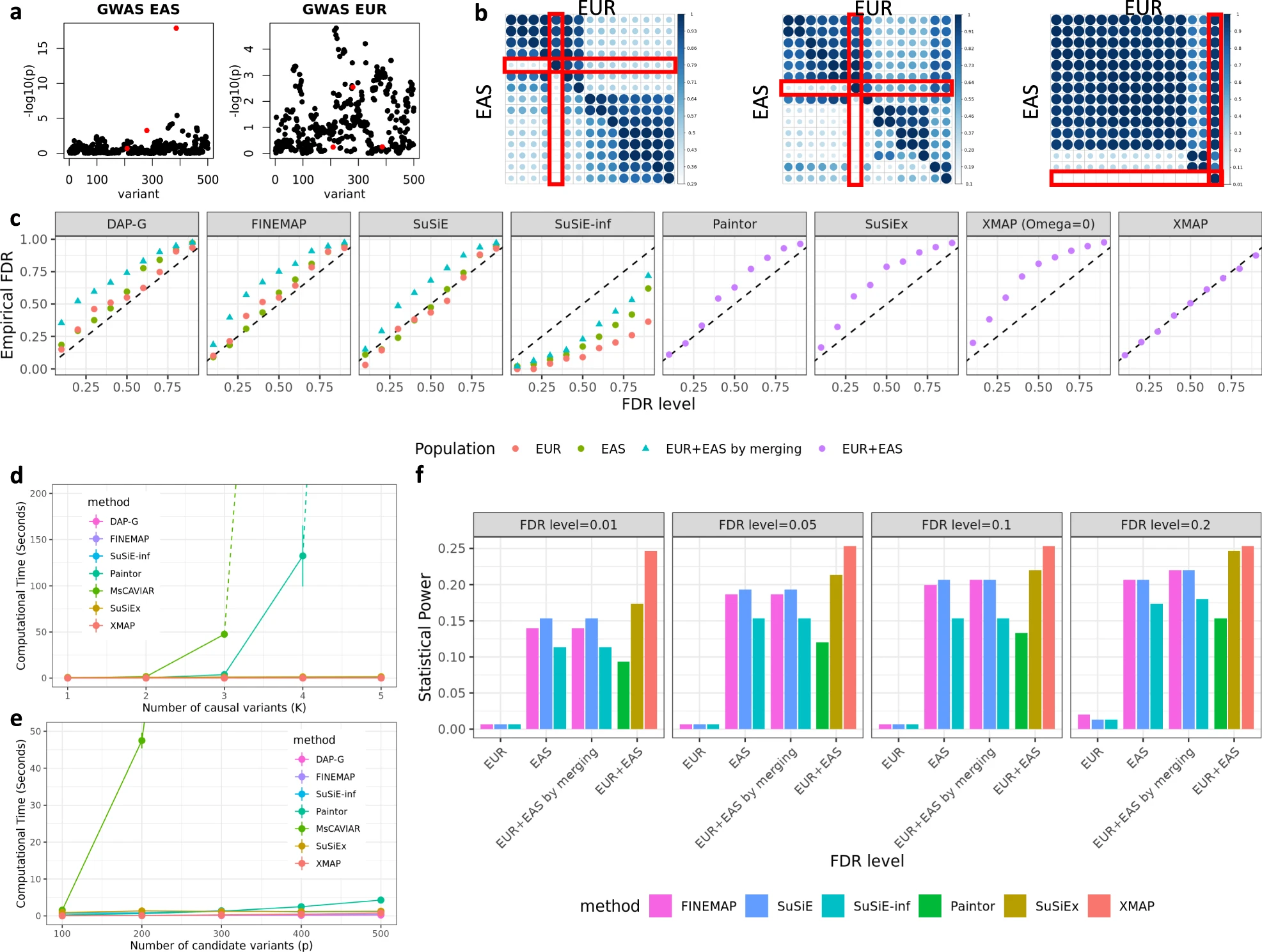
Realistic LD patterns in different populations: Genotypes of EUR samples from UKBB and genotypes of EAS samples from a Chinese cohort.
Benefit of leveraging genetic diversity: Region in chromosome 22 with p = 500 SNPs. Selected 3 candidate SNPs that: (i) In EUR population in high LD with at least three non-causal SNPs. (ii) In EAS population weakly correlated with non-causal SNPs.
Unbalanced populations samples: n2=20,000 samples from the EUR population and n1: 5000, 10,000, 15,000, and 20,000 from the EAS population.
Reference LD matrices: Used the EUR LD matrix estimated with 337,491 British UKBB samples and estimated the EAS LD matrix with 35,989 EAS samples from the Chinese cohort.
Scenarios:
Scenario 1: Demonstrated the benefit of cross-population fine-mapping by generating GWAS data without confounding bias.
Settings:
Polygenic effects generated for all 44,728 SNPs in chromosome 22 include the 500-SNPs target region. Total heritability: 5e-3.
Heritability of the target SNPs: 50 fold higher than the non-causal SNPs.
Effect sizes of the target SNPs are not necessarily the same in the two populations.
Genetic correlation between the two populations: 0.8.
Standardized X to have zero mean and unit variance.
50 simulation replicates.
Identified causal SNPs by controlling the global FDR.
Evaluation: FDR calibration, power, computational efficiency, and the robustness when mis-specifying the genetic effects.
Fig. 2: a Manhattan plots. b Heat maps showing the absolute correlations between the three causal SNPs and their nearby SNPs in two populations. c Comparisons of FDR control. d,e CPU timings. f Comparisons of statistical power.
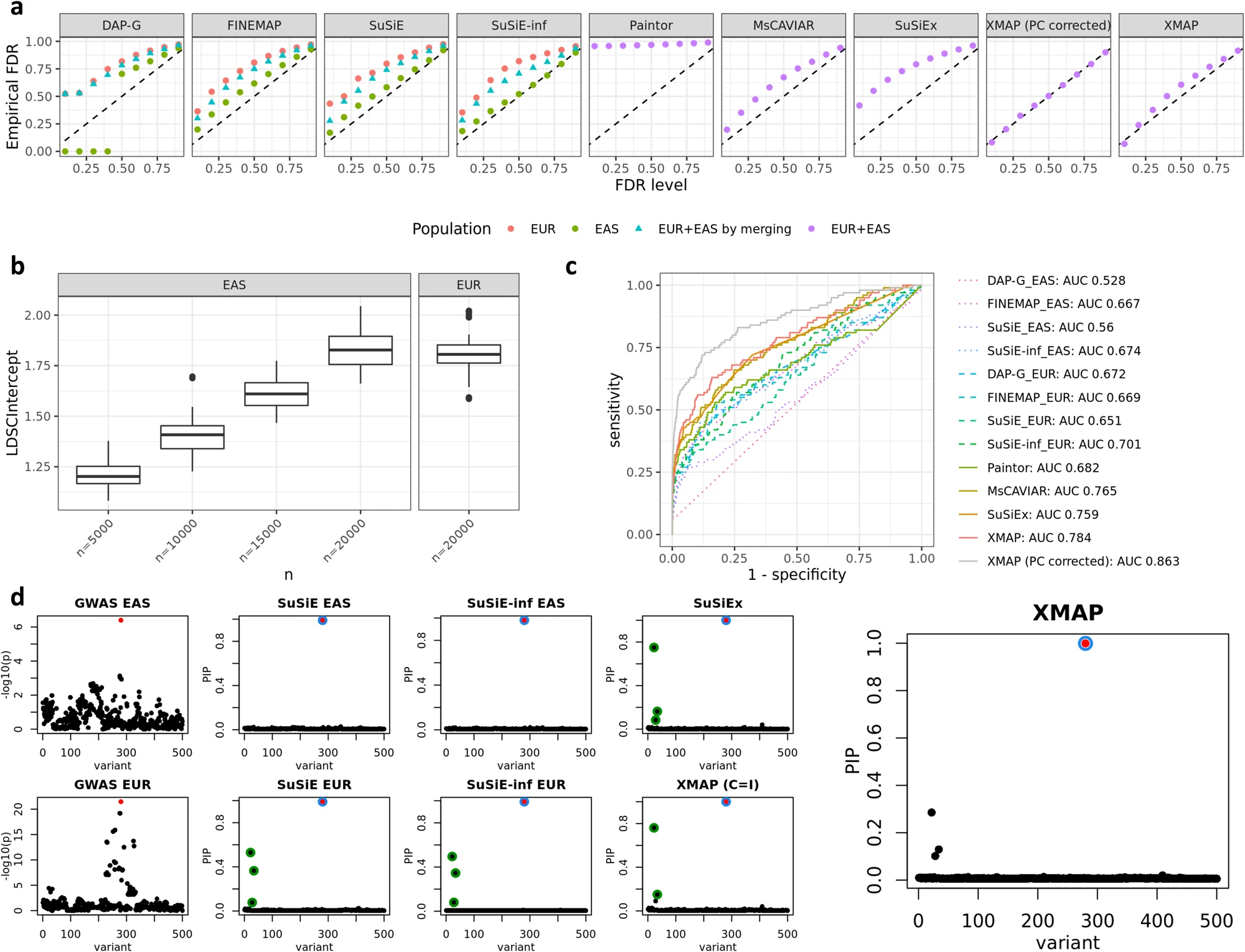
Scenario 2: Examined the effectiveness of XMAP in correcting confounding bias by simulating GWAS summary data with unadjusted sample structure.
Settings: Simulated unadjusted confounding bias with the first principal components from the two populations. Rescaled PC1 to have mean zero and variance 0.05 and PC2 to have mean zero and variance 0.2 which aim to introduce the proper level of inflation in the summary statistics.
Regress phenotype vectors on each SNP excluding the PCs as covariates, representing the scenario with unadjusted confounding bias.
Regress phenotype vectors on each SNP while including the PCs as covariates, representing the scenario with adjusted confounding bias.
Conclusion: It is effective to use the inflation constants to correct confounding bias in GWAS.
Fig. 3: a Comparison of FDR control. b Estimated LDSC intercepts. c Comparisons of ROC curves. d An illustrative example with a single causal signal.
Real Data Analysis
LDL GWASs:
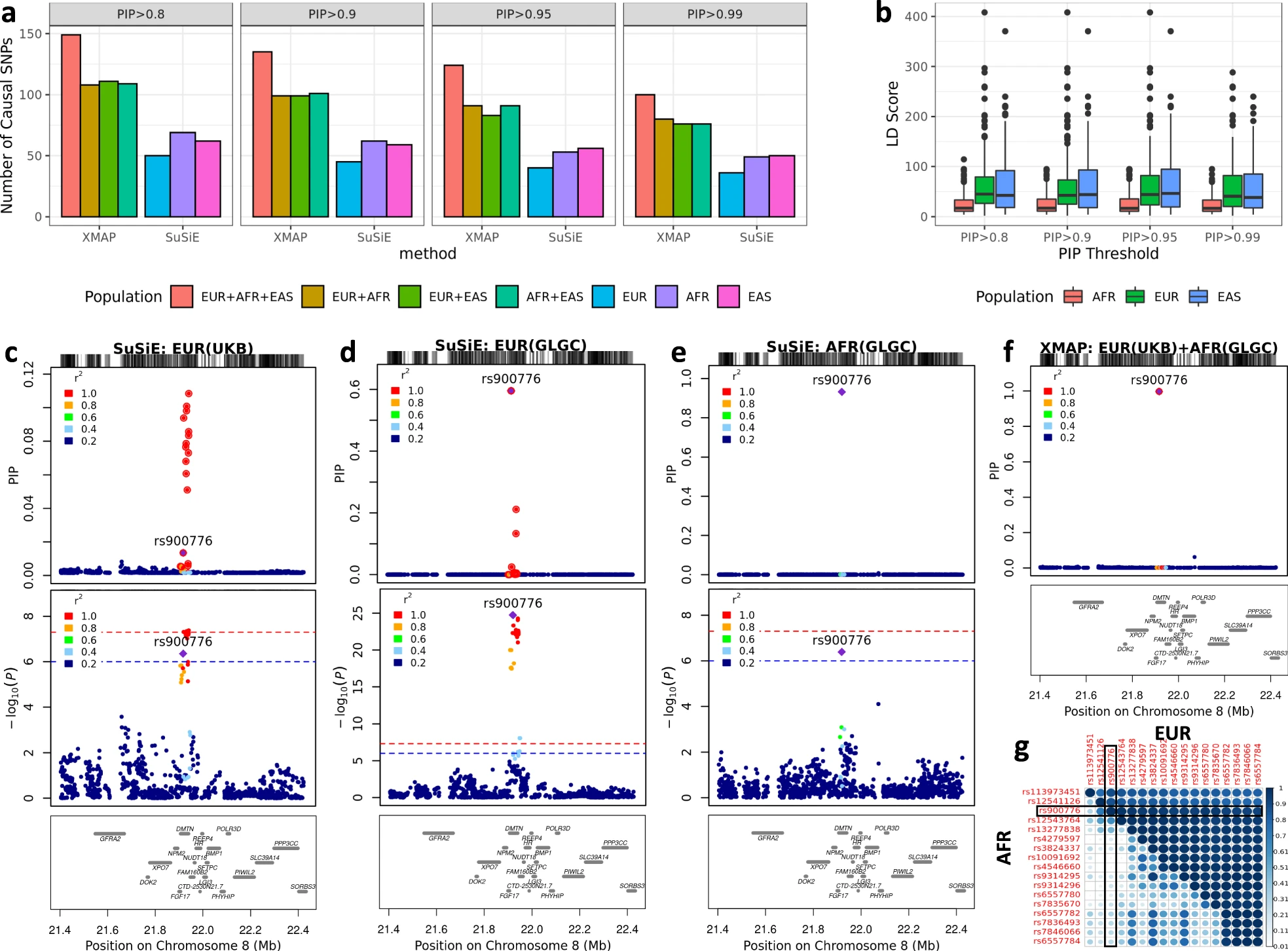
Data: GWASs of AFR and EAS (by GLGC) and EUR (by UKBB and GLGC).
For AFR, we estimated the LD matrices by using 3,072 African individuals from UKBB as reference samples.
Confounding bias: The LDSC intercepts estimated from all LDL GWASs were not substantially different from one, suggesting an ignorable confounding bias here.
Credibility: Evaluated the replication rate using an independent LDL GWAS from the EUR population.
Improvement of the power: Use rs900776 as an example to show the improvement of fine-mapping power and resolution by XMAP is owing to leverage the genetic diversity. [should compare the results of XMAP in 3 populations separately and altogether?]
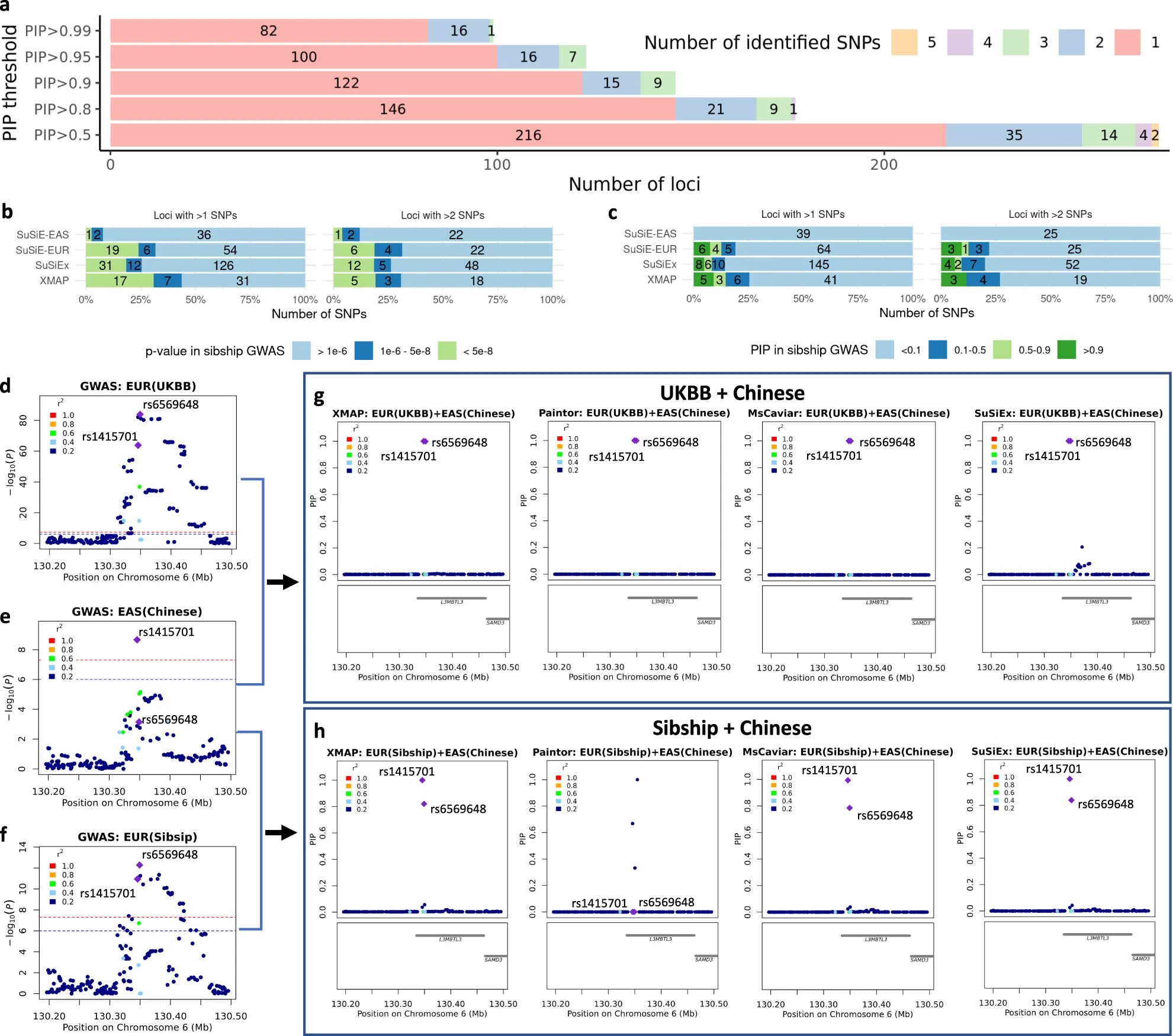
Fig. 4: a # causal signals identified by XMAP and SuSiE with different PIP thresholds. b The LD score distribution of putative causal SNPs identified by XMAP. c-f Fine-mapping of locus 21.4 Mbp–22.4 Mbp in chromosome 8. g Absolute correlation in EUR and AFR among the SNPs within the level-99% credible set. The SNP rs900776 is highlighted in the heat map.
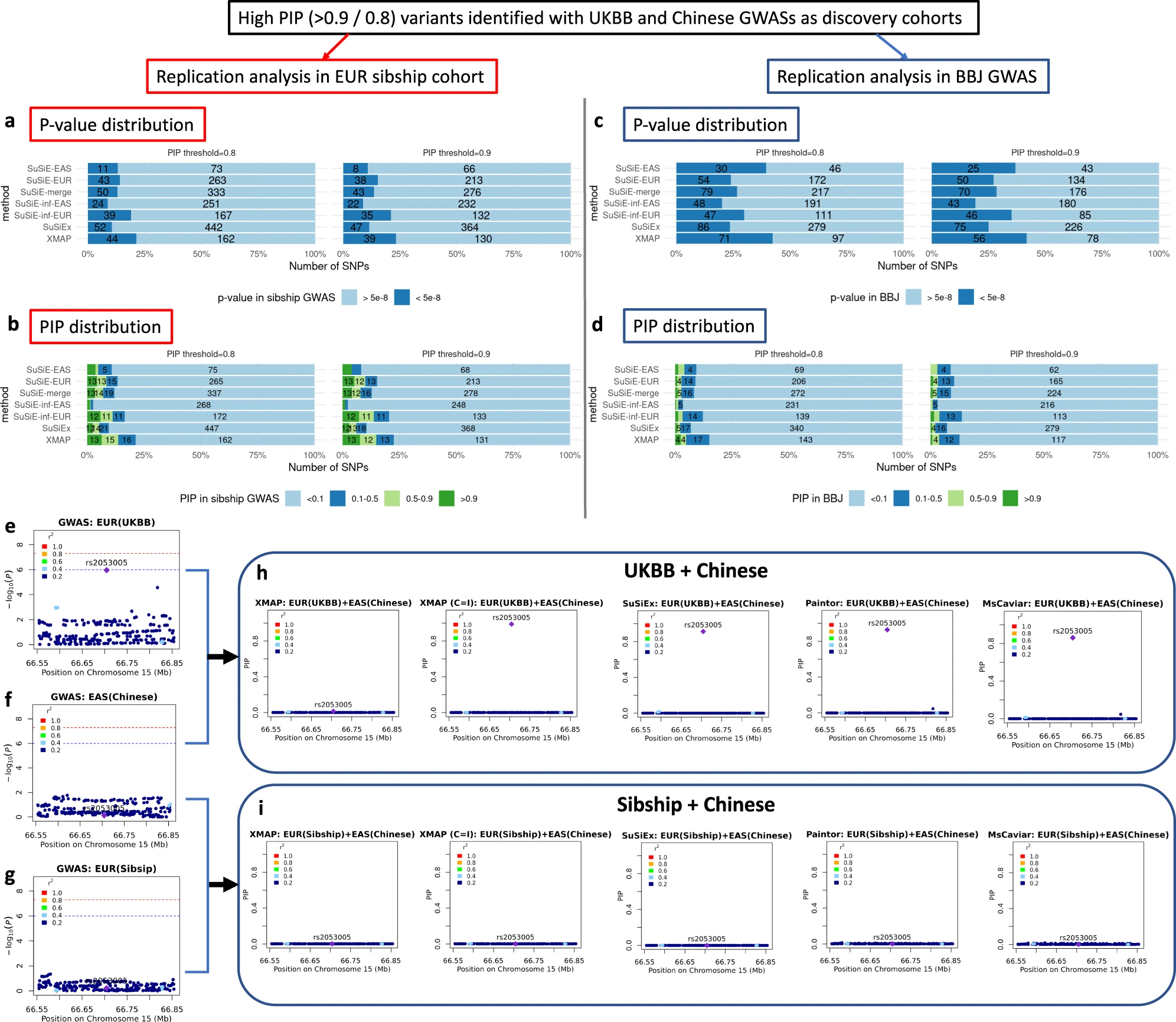
Height GWASs: which were well known to be affected by population structure.
Aim: Investigate the ability of XMAP in correcting confounding bias and reducing false positive signals.
Process: First applied fine-mapping methods to discovery GWAS datasets, and then evaluated the credibility in replication datasets from different population backgrounds.
Data: Strong confounding bias: EUR GWAS from UKBB and a Chinese GWAS.
Replication data: Ignorable confounding bias: Within-sibship GWAS from European population, which was known to be less confounded by population structure. The GWAS from BBJ cohort from EAS background.
Result: Rs2053005 could be a false positive and XMAP was able to exclude this signal by correcting the confounding bias.
Fig. 5: a-d Overview of replication analyses of high-PIP fine-mapped SNPs across populations: bar charts showing the fraction and number of fine-mapped SNPs with p-value < 5e−8 in the replication cohorts of EUR Sibship GWAS and BBJ cohorts and bar charts showing the distribution of PIP for fine-mapped SNPs computed by SuSiE in the replication cohorts of EUR Sibship GWAS and BBJ. e-i Fine-mapping of locus 66.55 Mbp–66.85 Mbp in chromosome 15.
Multiple causal signals: XMAP was able to identify multiple causal signals within a locus.
Data: Same with 2.
Results: XMAP, MsCaviar, and SuSiEx robustly identified multiple causal signals when the sample size decreased. XMAP robust to the choice of K which is max # of causal signals.
Fig. 6: a Distributions of the number of putative causal SNPs identified by XMAP under different PIP thresholds. b,c The p-value / PIP distributions in the Sibship GWAS replication cohort, threshold set as 0.9. d-h A demonstrative example using the locus 130.2 Mbp–130.5 Mbp in chromosome 6. Rs1415701 and rs6569648 had highly probability to be casual.
Single-cell data integration: XMAP results can be effectively integrated with single-cell datasets to identify disease/trait-relevant cells.
Data: Blood traits from scATAC-seq dataset that encompasses multiple hematopoietic lineages.
Process: ...
Result: Better interpretation of risk variants in their relevant cellular context, gaining biological insights into causal mechanisms at single-cell resolution.
Limitations
Assumptions: XMAP assumes that the causal variants are shared across populations, which may not be true for some signals (Same as PAINTOR and MsCAVIAR).
Disproportionate distribution of causal variants: Causal variants are reported to be distributed disproportionately in the genome, depending on the functional context of the genomic regions.
Gene-level effects: Gene-level effects can be more stably shared across populations, as compared to SNP-level effects. Leveraging the genetic diversity at the gene-level for fine-mapping can be an interesting direction.
Reference
Cai M, Wang Z, Xiao J, et al. XMAP: Cross-population fine-mapping by leveraging genetic diversity and accounting for confounding bias[J]. Nature Communications, 2023, 14(1): 6870.